ESSAY ON PAPER
An automated phylogenetic tree-based small subunit rRNA taxonomy and alignment pipeline (STAP).My first PLoS One paper .... yay: automated phylogenetic tree based rRNA analysis
Well, I have truly entered the modern world. My first PLoS One paper has just come out. It is entitled "An Automated Phylogenetic Tree-Based Small Subunit rRNA Taxonomy and Alignment Pipeline (STAP)" and well, it describes automated software for analyzing rRNA sequences that are generated as part of microbial diversity studies. The main goal behind this was to keep up with the massive amounts of rRNA sequences we and others could generate in the lab and to develop a tool that would remove the need for "manual" work in analyzing rRNAs.
The work was done primarily by Dongying Wu, a Project Scientist in my lab with assistance from a Amber Hartman, who is a PhD student in my lab. Naomi Ward, who was on the faculty at TIGR and is now at Wyoming, and I helped guide the development and testing of the software.
We first developed this pipeline/software in conjunction with analyzing the rRNA sequences that were part of the Sargasso Sea metagenome and results from the word was in the Venter et al. Sargasso paper. We then used the pipeline and continued to refine it as part of a variety of studies including a paper by Kevin Penn et al on coral associated microbes. Kevin was working as a technician for me and Naomi and is now a PhD student at Scripps Institute of Oceanography. We also had some input from various scientists we were working with on rRNA analyses, especially Jen Hughes Martiny
We made a series of further refinements and worked with people like Saul Kravitz from the Venter Institute and the CAMERA metagenomics database to make sure that the software could be run outside of my lab. And then we finally got around to writing up a paper .... and now it is out.
You can download the software here. The basics of the software are summarized below: (see flow chart too).
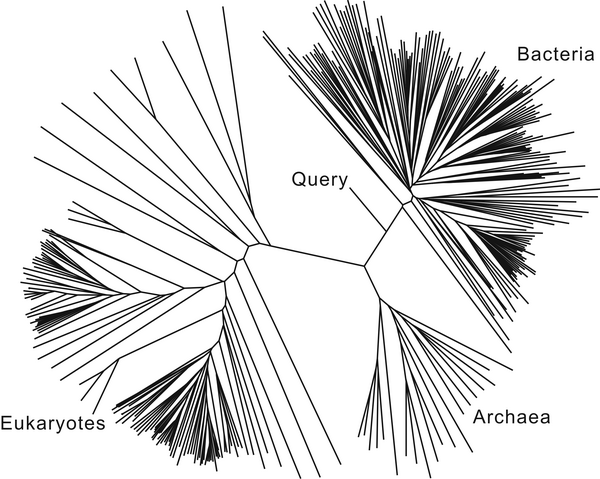
Stage 1: Domain Analysis
Take a rRNA sequence
blast it against a database of representative rRNAs from all lines of life
use the blast results to help choose sequences to use to make a multiple sequence alignment
infer a phylogenetic tree from the alignment
assign the sequence to a domain of life (bacteria, archaea, eukaryotes)
Stage 2: First pass alignment and tree within domain
take the same rRNA sequence
blast against a database of rRNAs from within the domain of interest
use the blast results to help choose sequences for a multiple alignment
infer a phylogenetic tree from the alignment
assign the sequence to a taxonomic group
Stage 3: Second pass alignment and tree within domain
extract sequences from members of the putative taxonomic group (as well as some others to balance the diversity)
make a multiple sequence alignment
infer a phylogenetic tree
From the above path, we end up with an alignment, which is useful for things such as counting number of species in a sample as well as a tree which is useful for determining what types of organisms are in the sample.
I note - the key is that it is completely automated and can be run on a single machine or a cluster and produces comparable results to manual methods. In the long run we plan to connect this to other software and other labs develop to build a metagenomics and microbial diversity workflow that will help in the processing of massive amounts of sequence data for microbial diversity studies.
I should note this work was supported primarily by a National Science Foundation grant to me and Naomi Ward as part of their “Assembling the Tree of Life” Program (Grant No. 0228651). Some final work on the project was funded by the Gordon and Betty Moore Foundation through grant #1660 to Jonathan Eisen and the CAMERA grant to UCSD.

COMMENTS