ESSAY ON PAPER
Stalking the fourth domain in metagenomic data: searching for, discovering, and interpreting novel, deep branches in marker gene phylogenetic trees.The story behind the story of my new #PLoSOne paper on "Stalking the fourth domain of life" #metagenomics #fb
Well, here goes.
This is a post about a paper that has been a long long time coming. Today, a paper of mine is being published in PLoS One. The paper is titled "Stalking the Fourth Domain in Metagenomic Data: Searching for, Discovering, and Interpreting Novel, Deep Branches in Marker Gene Phylogenetic Trees" and is available at http://dx.plos.org/10.1371/journal.pone.0018011. (or if that link does not work you can get a copy here). This paper represents something I started a long time ago and I am going to try to describe the story behind the paper here.
I note - we are not doing a press release for the paper, for a few reasons. But one of them is that, well, I am starting to hate press releases. So I guess this is kind of my press release. But this will be a bit longer than most press releases. I note - my key fear here is that somehow in my communications with the press or in our text in the paper or in this post I will overstate our findings. Here is the punchline - we found some very phylogenetically novel forms of phylogenetic marker genes in metagenomic data. We do not have a conclusive explanation for the origin of these sequences. They may be from novel viruses. The They may be ancient paralogs of the marker genes. Or they may be from a new branch of cellular organisms in the tree of life, distinct from bacteria, archaea or eukaryotes. I think most likely they are from novel viruses. But we just don't know.
UPDATE: Am posting some links here to news stories/blogs about our paper
- Glimpses of the Fourth Domain? by Carl Zimmer at his always stellar "The Loom"
- Biology's 'dark matter' hints at fourth domain of life by Colin Barras at the New Scientist
- 21st century science publishing will be multilevel and multimedia by PZ Myers
- German Radio Story
- Chinese news report
- PLoS One Blog Pick of the Month for March 2011 by Brian Mossop
- Brazilian news story from @info
- There might be at least one other, previously hidden, domain of life by Francis Martin
- Pronađen potpuno novi "domen" života
- Craig Venter's study of marine DNA finds new branches on the tree ... Telegraph article #1
- Scientist finds a whole new 'domain' of life - Telegraph article #2
- Scientist stumbles on new class of living things - Zee News
- A new domain of life: Plenty more bugs in the sea from The Economist
- DNA Analysis Hints At a Fourth Domain of Life - Slashdot
First - a summary of what we did.
In the paper, we searched through metagenomic data (sequences from environmental samples) for phylogenetically novel sequences for three standard phylogenetic marker genes (ss-rRNA, recA, rpoB). We focused on sequences from the Venter Global Ocean Sampling data set because, well, we started this analysis many years ago when that was the best data set available (more on this below). What we were looking for were evolutionary lineages of these genes that were separate from the branches that corresponded to the three known "Domains" of life (bacteria, archaea and eukaryotes).
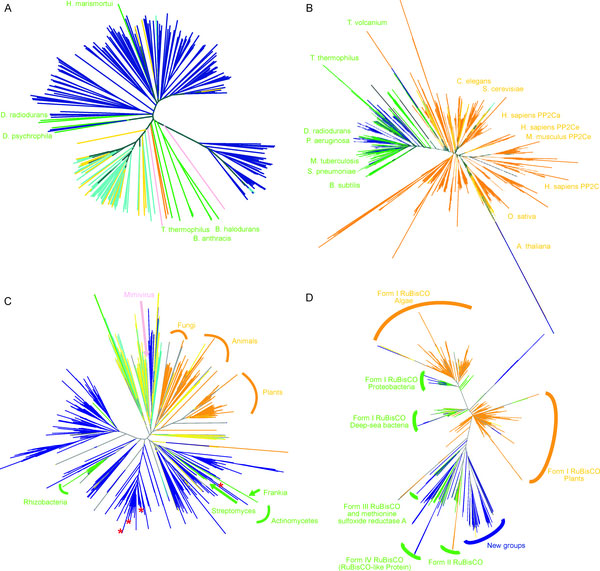
To search for such novel lineages in the metagenomic data, we built evolutionary trees using these genes where we included sequences from known organisms (and viruses) as well as sequences from metagenomic data. We then looked through the trees for groups that were both phylogenetically novel and included only environmental data (i.e., they were new compared to known organisms or viruses). This method did not work very well for rRNA sequences (largely because making high quality alignments of short phylogenetically novel rRNA sequences was difficult - more on this below). But with RecA and RpoB homologs we were able to generate what we believe to be robust phylogenetic trees. And in these trees we found evidence for phylogenetically very novel sequences in environmental data.
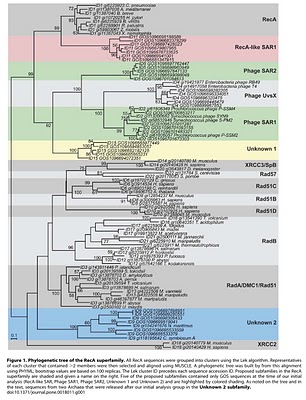
Figure 1. Phylogenetic tree of the RecA superfamily.
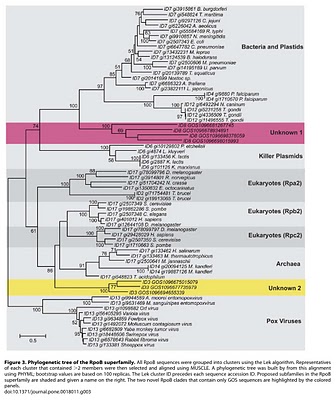
Figure 3. Phylogenetic tree of the RpoB superfamily.
We then propose and discuss four potential mechanisms that could lead to the existence of such evolutionarily novel sequences. The two we consider most likely are the following
- The sequences could be from novel viruses
- The sequences could be from a fourth major branch on the tree of life
Unfortunately, we do not actually know what is the source of these sequences. So we cannot determine which of the theories is correct. Obviously if there is a novel lineages of cellular organisms out there, well, that would be cool. But we have no evidence right now if that is what is going on. Personally, I think it is most likely that these novel sequences are from weird viruses. But as far as we can tell, they truly could be from a fourth major branch of cellular organisms and thus even though we did not have the story completely pinned down, we decided to finally write up the paper to get other people to think about this issue.
Below I give all sorts of other details about the project in the following areas
- The history of the project
- More detail on what is in the paper
- Follow up analysis and rapid posting with google Know
- Data deposition in Dryad
- Who was involved
- UPDATE: Funding for this work
The history of the project
Well, this is one of those projects for which the history is hard to explain. We started this work in 2004 when I was helping Venter and colleagues analyze the Sargasso Sea metagenome data. I was working at TIGR in 2003, which are the time was a sister institute to some of the institutes affiliated with the J. Craig Venter Institute (JCVI) (it was a complicated time). Craig had led a project to do a massive amount of shotgun sequencing of DNA isolated from the Sargasso Sea, which had been the site of many previous studies of uncultured microbes. And Craig, as well as some of the people working with him including John Heidelberg who was at TIGR, had asked me to help in analysis of the data. So I eventually went to a meeting about the project and got involved. It was quite exciting and I put a lot of effort into helping analyze the data.
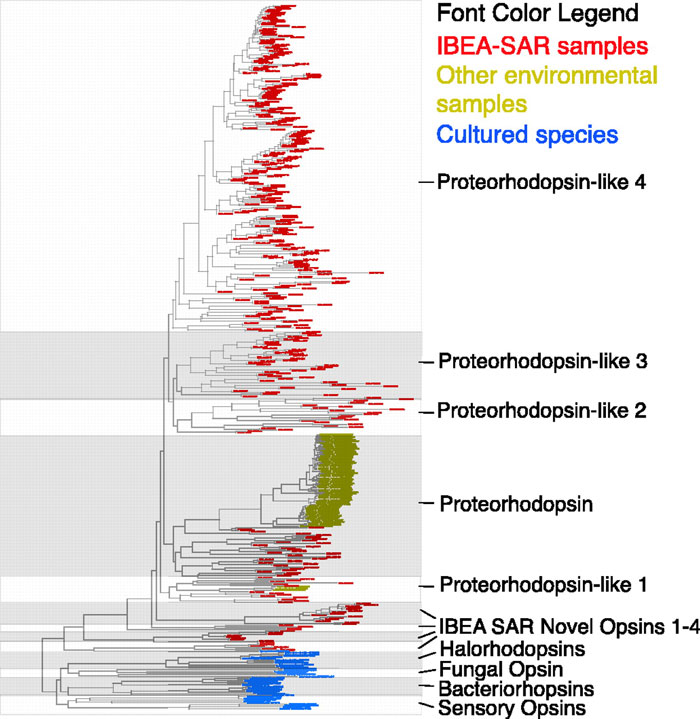
As part of my work on the project, I and Martin Wu and Dongying Wu did a variety of phylogenetic studies of genes and gene families. One of these, was a phylogenetic analysis of proteorhodopsin homologs showing massively more diversity in the Sargasso data than in the PCR experiments done by Delong and Beja and others.
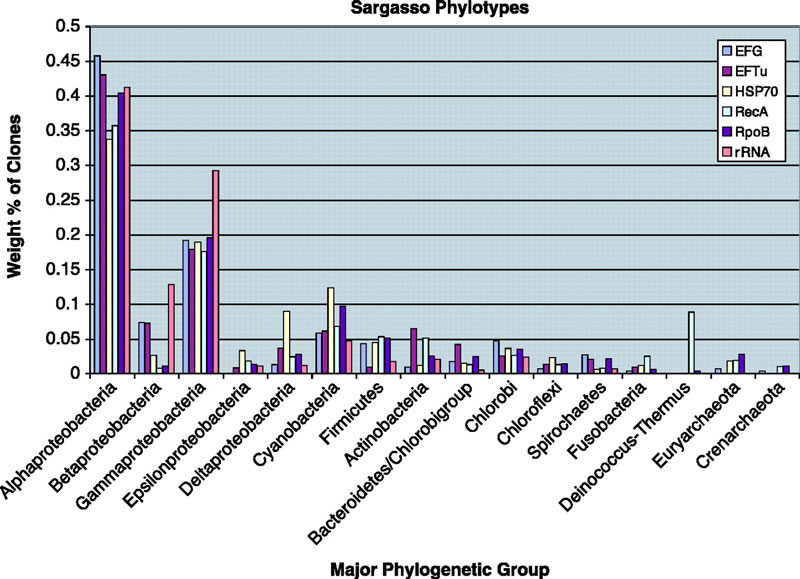
We also did the first "phylotyping" in metagenomic data using genes other than rRNA. We built trees of bacterial ss-rRNAs, RecAs, RpoBs, HSP70s, EF-Tus and EF-Gs and then assigned each sequence to a phylum from the trees. In this analysis we found a variety of interesting things.
One thing I did not include in the Sargasso paper was an analysis I did of RecA homologs where I tried to include ALL RecA-like genes from bacteria, archaea, eukaryotes and viruses. The trees I made were a bit unusual but I was not sure that the alignments I had made were robust or that I had found all the RecA-like genes of interest so I did not even show this to Craig et al. at the time.
UPDATE: I note - our work on this project was supported by a grant from the NSF Assembling the Tree of Life program that was awarded to me and Naomi Ward and Karen Nelson. Those funds supported the development of many of the informatics tools we used in this analysis and Martin and Dongying were both working on that project.
After the Sargasso paper was published in 2004 though, I continued to fester about the RecA trees. And I wondered - if instead of trying to classify bacterial sequences into phyla, what if I tried to look for RecAs, rRNAs and other genes that were completely new branches in the tree of life? I got the chance to start to play with this concept again when Venter and crew asked me to help analyze the data coming out of the Global Ocean Sampling project. Again, this project was very exciting and interesting.
As part of the project, I helped Shibu Yooseph and others look into whether the GOS data revealed any completely new types of functionally interesting genes, much like I had shown for proteorhodopsin in the Sargasso data.
And again my mind started wandering towards the question of "OK - so - if there are all these very unusual and novel functionally interesting genes, what about looking for unusual and very novel phylogenetic marker genes"? So finally, I got back to work on the issue.
And so I built a better RecA tree by first pulling out all possible homologs of RecA and RecA like proteins from the GOS data and then building an alignment and a tree. And there they were. Some very f*%&$ novel RecAs - distinct from any previously known RecA like proteins as far as I could tell. And so with help from Dongying and the JCVI crew, we started building a story about novel RecAs. And then we looked at RpoBs. And found novel ones too. And in mid 2006 while Shibu and Doug worked on their papers that were to be submitted to PLoS Biology and I worked on a review paper too, I told Emma Hill (who has since changed her name to Emma Ganley due to some sort of wedding thing) at PLoS Biology about the an analysis that was consistent with the existence of a fourth domain of life. No overstating our findings really - just that we found very novel phylogenetic marker genes. And that I was working on a paper on it. But alas I never got it done, though I was happy to have convinced Venter to send the GOS papers to PLoS Biology and I think the papers that came out were good. Among the papers were my review (Environmental Shotgun Sequencing: Its Potential and Challenges for Studying the Hidden World of Microbes, Doug Rusch's diversity paper The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific and Shibu's protein family paper The Sorcerer II Global Ocean Sampling Expedition: Expanding the Universe of Protein Families as well as many others as part of the Ocean Metagenomics Collection at PLoS.
And in the midst of all of this, we had our first child and we wanted to move back to Northern California to be closer to family (my wife's family is all in the Bay Area and my sister and brother Michael were in N. Cal too). So I applied for jobs and eventually took at job at UC Davis and we moved to Davis. Needless to say, all of that put a bit of a crimp in my work productivity. And once I was up and running at Davis, it just took a long time to get back to the searching for novel deep branches in the tree of life. But finally, we did it (with periodic prodding from Craig Venter). And we put together a paper and got it submitted to PLoS One in October. The reviews were very positive and enormously helpful. And we finally got a revision in January and it was officially accepted in February 2011. Only some seven years after my first work on the project. Whew.
More detail of what is in the paper
Well, I am going to be posting here some additional detail on what is in the paper.
Why we punted on analysis of very novel rRNAs.
The problem with rRNA is that the sequences that come from environmental samples are not complete (i.e. they only correspond to portions of the rRNA genes). Unfortunately, this makes a key step in phylogenetic analysis difficult - the alignment of sequences. We actually found about 200 rRNA sequences that seemed unusual in a phylogenetic sense. However, we were not convinced that the alignments of these fragments to other rRNAs was robust. This is because the alignment of rRNAs is best done making use of the base pairing secondary structure of the molecule and not the base sequence (i.e., primary structure).
With only rRNA fragments, we could not use the secondary structure to do the alignments because you need to whole molecule to determine the best folding. Combined with the fact that we were searching for very distantly related ribosomal RNAs which would be hard to align even if we had the whole molecule, we were stuck for a bit. It seemed impossible to look for really novel organisms.
So that is when we turned to other genes. The key for this is that there are protein coding genes that are universal and that for known organisms show similar patterns to rRNA in trees. In fact, in 1995 I wrote a paper showing that trees of RecA were very similar to trees of rRNA. RpoB is also considered a very robust phylogenetic marker. For organisms that we have in the lab (i.e., cultured) - many people use these other genes for phylogenetic analysis. rRNA has been very important in part because of the ease with which one can PCR amplify it from environmental samples and the fact that it is very hard to PCR amplify protein coding genes from the environment. Metagenomics changes this. With random sequencing, you get data from all genes. This means we can pick and choose genes to analyze for phylogenetic analysis and do not have to rely on rRNA.
So we went after RecA first, because it has been shown to be a good phylogenetic marker for studies of the tree of life. And we found some very novel branches in the RecA tree. And after analyzing these and convincing ourselves that they were indeed phylogenetically very novel we went after RpoB. And also found very novel branches.
So the phylogenetic analysis I think is very robust.
RecA and RpoB as phylogenetic markers
Many genes have been used as alternatives to rRNA genes to build "Trees of Life" including all organisms. Each has their own flavors of advantages and drawbacks. Two commonly used ones are the RecA and RpoB superfamilies.
The many possible explanations for finding novel forms of phylogenetic marker genes
The phylogenetically novel phylogenetic marker genes we found could have many explanations including that they could be ancient paralogs of these genes (but not found in any genomes we have available), they could be from viruses, or they could be from a novel branch on the tree of life. Or our trees could be bad. We think the latter is somewhat unlikely as our analysis has many lines of support. For example our RecA trees are very similar to those from a comprehensive study from M. Nei's lab except they did not include the metagenomic data. But I guess it is still a possibility that our trees are biased in some way (e.g., by long branch attraction or bad alignments)
Follow up analysis and rapid posting via Google Knol
Amazingly and a bit sadly, I think we rushed the paper out. We left out one thing partly by accident - we had done an analysis of the locations from which these novel RecA and RpoB sequences had come. And somehow, in our final push to get the paper out, we left this out. I will be posting this information as soon as possible here and on the PLoS One site.
In addition, after submitting the revision of our paper, we realized that we might be able to do a deeper analysis on one aspect of the work - how RpoB homologs from unusual DNA viruses compared to our novel sequences. We had included some RpoBs from DNA viruses in our analyses but not all that were available. So Dongying Wu did a very rapid additional analysis, adding some additional RpoB homologs to our alignment and making a tree of them. We then wrote a Google Knol about this new tree and submitted the Knol to PLoS Currents "Tree of Life" where it is currently in review. We are publishing the preprint of this Knol to make it available to all even while it is in review.
Data availability
There is a move afoot to make sure all data/tools associated with publications are readily available. We used publicly available sequence data and as much as possible publicly available tools for our work . We are trying to release as much as possible to allow people to re-analyze our work and to do any of the work themselves. We have therefore made use of the Dryad Data deposition service to post some of this material (see http://datadryad.org/handle/10255/dryad.8385).
Who was involved
- Dongying Wu a brilliant "Project Scientist" in my lab led the project (Project Scientist is one of the UC positions that is like what others call "Senior Scientist"). Dongying is simply one of the best bioinformaticians/computational biologists I have ever met. He was first author on many key papers from my lab including the Genomic Encyclopedia paper that came out last year and the glassy winged sharpshooter symbionts paper that came out a few years ago. Dongying worked in my group at TIGR and moved with me to UC Davis and currently splits his time between UC Davis and the DOE Joint Genome Institute.
- Martin Wu. Martin is an Assistant Professor at the University of Virginia. Prior to that he was a Project Scientist in my lab at Davis and a post-doc in my lab at TIGR. He is also a phenomenal bioinformatician / computational biologist. He developed the AMPHORA software in my lab and also led many genome projects (back when sequencing a genome was hard ...) including that of the first Wolbachia genome and that of a very unusual bug Carboxydothermus hydrogenoformans. Martin helped with some of the genome analyses as part of this work.
- Aaron Halpern, Doug Rusch and Shibu Yooseph are all bioinformaticians from the J. Craig Venter Institute (Aaron is no longer there). All three helped with different aspects of dealing with and analyzing the GOS data and all three have been remarkably patient as this work dragged on and on.
- Marv Frazier from the JCVI was helpful in the initial set up and conceptualization of the project.
- J. Craig Venter is, well, Craig Venter, and he was involved in multiple aspects of the project including thinking about how and where to look for unusual sequences and interpreting some of the results.
UPDATE: Funding for this work
Most of my labs early work on this project was supported by a grant we had from the Assembling the Tree of Life program at the National Science Foundation (grant 0228651 to me and Naomi Ward). In that project we were working on sequencing and analyzing genomes from phyla of bacteria for which genomes were not available at the time. As part of this work we were designing methods to build phylogenetic trees from metagenomic data because we thought that our new genomes would be very useful in helping analyze metagenomic reads and figure out from which phyla they came. Later work on the project was supported by a grant to me, Jessica Green and Katie Pollard from the Gordon and Betty Moore Foundation (grant 1660).
Some questions that might be asked and some answers (based in part on questions I have gotten from reporters). Note if you have other questions please post them here or on the PLOS One site for the paper.
- Why no press release? Well, in part, because I sent information too late (shocking I know) to the Davis Press Office. But also because they have gotten suddenly busy with some Japan earthquake related actions. But also because, well, I really hate a lot of press releases. And finally, my brother had dinner with Carl Zimmer recently and apparently they discussed the possibility of having no press releases associated with papers. So here goes ....
- Really - what took so long? I would like to say the US Government made us hold back on publishing this until they could look into whether Venter collected ocean data from Roswell, NM or not. But really, the story above is true. We just did not get it done earlier.
- Why do you not know the source of the DNA (i.e., cells, viruses, etc)? This is why there was a six year wait between discovery and writing this up. We kept thinking we would be able to find the organisms but since I moved from TIGR and started a new job, we just never got around to getting to the source. We therefore decided to open this up to others who will hunt for the source by writing up the paper.
- Why did you not rename the Unknown 2 group in the RecA tree? We should have renamed our group "Thaumarchaeota" or something like that. When we did the initial analysis our group was novel. And then a few years ago a few groups obtained data from what is thought to be the third major lineage of Archaea - referred to by some as Thaumarchaeota. This is to go with the Euryarchaeota and Crenarchaeota. See http://www.ncbi.nlm.nih.gov/pubmed/20598889 for example.
- One of the clades in the RecA tree (XRCC2) seems out of place phylogenetically. I can see how that is confusing. The XRCC2 clade is very weird and hard to figure out. It is not the "normal" eukaryotic genes - those are the Rad51/DMC1 genes. One complication with the RecA family is that there have been duplication events to go with the species evolution. And thus eukaryotes have Rad51, DMC1, Rad51B, Rad51C, Rad57, XRCC3 and XRCC2. We tried to figure out where the XRCC2 group should go but it just was hard to place. The statistical support for its position (we used a method called bootstrapping) is low (note the lack of a number on the node where the branch leading to XRCC2 connects to the base of the tree). Most likely that group should be placed with some of the other eukaryotic groups. However, it seems likely that there was a duplication in the lineage leading up to the ancestor of eukaryotes and archaea (some studies have indicated they share a common ancestor to the exclusion of bacteria). Such a duplication would explain why basically all archaea have a RadA and and RadB and all / most eukaryotes have multiple paralogs as well.
- The Unknown 1 group in the
RpoBRecA tree seems to group with phage. What can you say about that? We think unknown 1 is potentially of viral origin but still cannot tell. The fact that is clusters with RecA superfamily members from phage suggests this but it is distant enough from known phage for us to not be confident in any predicted origin. As for derivative forms vs. independent branch - this is one of the big questions about viruses these days. Many viruses encode homologs of "housekeeping" genes found across bacteria, archaea and eukaryotes. And in many cases the viral versions of these genes appear to phylogenetically very novel. This is why the people studying mimivirus (which we refer to) suggest some viruses may in fact represent a fourth branch on the tree of life. It is possible that some viruses are in fact reduced forms of what were once cellular organisms - akin to parasitic intracellular species of bacteria possibly. - Why are these phylogenetically novel sequences so low in abundance? This is a key question. I think it would be easy to come up with a theory for these being rare or these being common. They might be rare if their niche is very limited today. Or they might be rare because they could not be very competitive with other organisms. Or they could be rare because they require some unusual interactions with other taxa. In addition, we have only looked carefully at ocean water samples. If these are common somewhere else (e.g., hotsprings, deep subsurface, etc) we would not yet have figured that out. We are looking at additional metagenomic data right now to see fi we can find any locations where relatives of these genes are more common
Some related papers by others worth looking at
- Can identification of a fourth domain of life be made from sequence data alone, and could it be done on Mars?
- Mimivirus: the emerging paradox of quasi-autonomous viruses.
- New evolutionary frontiers from unusual virus genomes
- Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4 domain of life including giant viruses.
- Giant viruses, giant chimeras: the multiple evolutionary histories of Mimivirus genes.
Some related papers by me possibly worth looking at
- The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific.
- The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families.
- Environmental genome shotgun sequencing of the Sargasso Sea.
- Unsuspected diversity among marine aerobic anoxygenic phototrophs.
- Environmental shotgun sequencing: its potential and challenges for studying the hidden world of microbes.
- A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea.
- A simple, fast, and accurate method of phylogenomic inference.
- An automated phylogenetic tree-based small subunit rRNA taxonomy and alignment pipeline (STAP).
- PhylOTU: a high-throughput procedure quantifies microbial community diversity and resolves novel taxa from metagenomic data.
- Introducing W.A.T.E.R.S.: a workflow for the alignment, taxonomy, and ecology of ribosomal sequences.
Some related blog posts I have written over the years
- Here's hoping molecular classification/systematics of cultured & uncultured microbes wins #NobelPrize in medicine
- Calling all phylogeneticists - we need your help with metagenomic data
- Scientist Reveals Secret of the Ocean: It's Him

COMMENTS